
Cybersicherheit für eingebettete Systeme
Von Medizinprodukten und intelligenten Fahrzeugen bis hin zu Industriecontrollern und Unterhaltungselektronik – eingebettete Systeme sind allgegenwärtig und zunehmend vernetzt. Doch […]

Join us at Qt C++ Warsaw Meetup - 21.08.2025
Sign up for free!
Verifikation und Validierung (V&V) sind zwei Grundpfeiler, die sicherstellen, dass ein Medizinprodukt sicher, wirksam und konform ist, bevor es überhaupt Patienten erreicht. Der Verifikations- und Validierungsprozess findet über den gesamten Produktentwicklungszyklus hinweg statt – von der ersten Entwurfsphase über die Produktion bis hin zu Änderungen nach dem Inverkehrbringen – und dient als formelle „Kontrollpunkte“, um Qualität und Leistung zu bestätigen. Kurz gesagt: V&V ist nicht nur eine regulatorische Pflichtaufgabe – es ist ein grundlegender Bestandteil des Risikomanagements, der Qualitätssicherung und des Testprozesses, der sowohl Anwender als auch Unternehmen schützt und gleichzeitig die Einhaltung gesetzlicher Vorschriften gewährleistet.
In diesem Artikel erfahren Sie:
Bei Scythe Studio unterstützen wir Teams für Medizinprodukte dabei, qualitativ hochwertige und normkonforme Software durch rigorose Tests und Validierung bereitzustellen. Egal, ob Sie Embedded-Firmware oder mobile Gesundheitsanwendungen entwickeln – wir helfen Ihnen, Ihre V&V-Testprozesse für Medizinprodukte zu stärken und regulatorische Risiken zu minimieren – und das bei gleichzeitiger Unterstützung eines effizienten Produktdesigns.
Sprechen wir darüber, wie Sie Ihr nächstes Produkt sicher, testfähig und auditbereit gestalten können.
Für Unternehmen der Medizintechnik und ihre Kunden steht viel auf dem Spiel, wenn V&V nicht korrekt durchgeführt wird. Verifikation und Validierung schaffen Vertrauen, dass ein Gerät über seine gesamte Lebensdauer hinweg korrekt und sicher funktioniert – mit direkter Auswirkung auf die Patientensicherheit. Ohne ein systematisches Prüfverfahren gegen die technischen Gerätespezifikationen und Benutzeranforderungen können Entwickler nicht sicher sein, dass das fertige Produkt:
Im Wesentlichen steht V&V im Zentrum der Qualitätsstandards und Vorschriften in der Medizintechnik, da diese Aktivitäten Konstruktionsmängel oder Leistungslücken erkennen, bevor sie Patienten schaden oder kostspielige Rückrufe verursachen. Der Designvalidierungsprozess ist entscheidend, um zu bestätigen, dass ein Gerät in realen Szenarien wie vorgesehen funktioniert.
Verifikation und Validierung verfolgen ähnliche, aber unterschiedliche Ziele zur Sicherstellung der Produktqualität. Verifikation fragt: „Haben wir das Produkt richtig gebaut?“ – und bestätigt, dass das Gerät gemäß den Spezifikationen konstruiert wurde. Validierung fragt: „Haben wir das richtige Produkt gebaut?“ – und bestätigt, dass das fertige Produkt die Benutzeranforderungen erfüllt und im realen Einsatz funktioniert. Beide Aspekte sind unerlässlich. Ein Gerät kann alle Verifikationstests bestehen (alle technischen Spezifikationen erfüllen) und dennoch bei der Validierung scheitern, wenn es das tatsächliche Problem des Nutzers nicht löst oder im Feld unsicher bzw. unpraktisch ist. Gründliches V&V ist daher der Weg, wie Hersteller kostspielige Neuentwicklungen verhindern, regulatorische Risiken reduzieren und sicherstellen, dass Geräte im realen Einsatz zuverlässig sind. Gut dokumentierte Testergebnisse sind entscheidend für den Nachweis der Konformität und für zukünftige Audits.
Aus Sicht des Lebenszyklus sind V&V-Aktivitäten als formelle Kontrollpunkte in den Designprozess eingebettet (oft im Zusammenhang mit Designkontrollen). Die Verifikation erfolgt häufig iterativ während der Entwicklung – durch das Testen von Prototypen medizinischer Geräte oder Komponenten im Vergleich zu den Designanforderungen – während die Validierung an produktionsgleichen Geräten (oder dem finalen Design) erfolgt, um zu beweisen, dass das Produkt für den Endnutzer funktioniert. Verifikation findet früh in der Entwicklungsphase statt, Validierung typischerweise später, mit den finalen Produktionseinheiten. Verifikation prüft, ob die Designausgänge den Gerätespezifikationen entsprechen. Validierung bestätigt, ob das Produkt den vorgesehenen Zweck erfüllt.
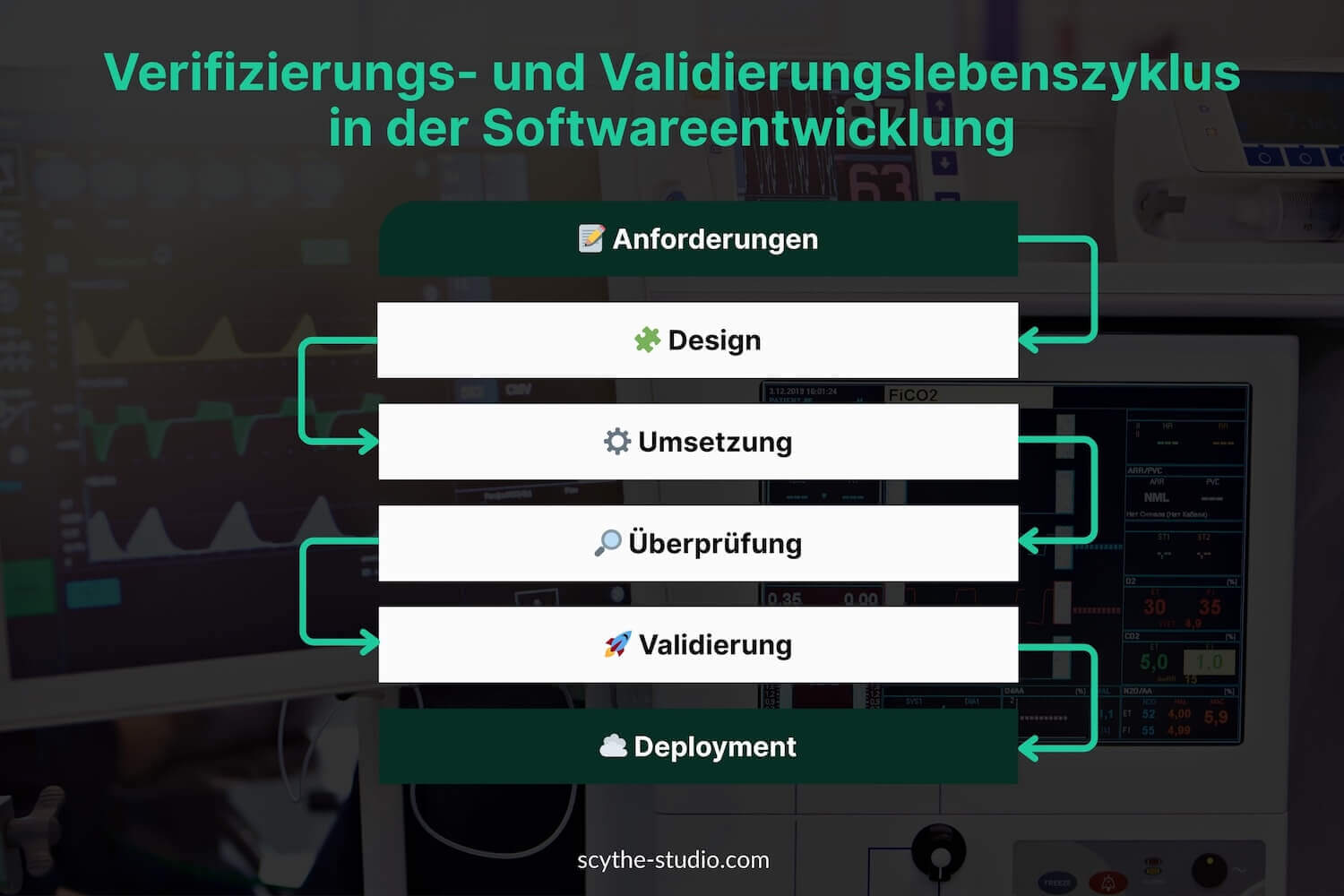
Regulierungsbehörden verlangen Nachweise für beide Aspekte, bevor ein Produkt zugelassen oder auf den Markt gebracht werden kann. In den USA wird die FDA ein Gerät (510(k), PMA usw.) nicht freigeben, ohne dass dokumentiert ist, dass die Designausgänge gegen die Eingaben verifiziert wurden und dass das Endgerät validiert wurde, um den Benutzeranforderungen zu entsprechen. In Europa erwarten Benannte Stellen bei der Prüfung der Technischen Dokumentation im Rahmen der EU-MDR umfassende V&V-Ergebnisse (einschließlich klinischer Nachweise), die die Einhaltung der allgemeinen Sicherheits- und Leistungsanforderungen belegen. Kurz gesagt: V&V ist das Qualitätstor, das jedes Medizinprodukt passieren muss, um sicherzustellen, dass es nicht nur korrekt gebaut ist, sondern auch die richtige Lösung für Patienten darstellt.
Es ist leicht, Verifikation und Validierung zu verwechseln, da beide Tests beinhalten und objektive Nachweise liefern. Dennoch beantworten sie grundlegend unterschiedliche Fragen. Eine klassische Eselsbrücke lautet:
Formal definieren die FDA und ISO dies wie folgt: Die Designverifikation ist die „Bestätigung durch Untersuchung und Bereitstellung objektiver Nachweise, dass festgelegte Anforderungen erfüllt wurden“. In der Praxis beziehen sich diese „festgelegten Anforderungen“ auf die Design-Eingabedaten – also die technischen Spezifikationen, die aus den Benutzerbedürfnissen abgeleitet wurden. Verifikationsaktivitäten beantworten die Frage: Haben wir jede Anforderung korrekt im Design umgesetzt? Wenn beispielsweise eine Designvorgabe lautet: „Der Katheter muss einem Berstdruck von mindestens 200 psi standhalten“, würde die Verifikation das Testen des Katheters auf Berstdruck umfassen, um diese Anforderung zu bestätigen. Jeder Designausgang (Zeichnungen, Prototypen, Code usw.) wird überprüft, um sicherzustellen, dass er den Eingabeanforderungen entspricht.
Designvalidierung hingegen ist die „Bestätigung durch Untersuchung und Bereitstellung objektiver Nachweise, dass die besonderen Anforderungen für einen bestimmten vorgesehenen Gebrauch konsistent erfüllt werden können“. Weniger formal fragt die Validierung: Erfüllt das gebaute Gerät die Bedürfnisse der Benutzer und den medizinischen Verwendungszweck in der realen Welt? Dies umfasst das Testen des Endprodukts (oder produktionsgleicher Einheiten) unter tatsächlichen oder simulierten Einsatzbedingungen, um sicherzustellen, dass das Gerät für den vorgesehenen Zweck geeignet ist. Beispielsweise bedeutet dies, zu validieren, dass ein Katheter tatsächlich zu besseren Patientenergebnissen in einem klinischen Umfeld beiträgt und für Fachpersonal benutzerfreundlich ist – also nicht nur, dass er technischen Spezifikationen entspricht, sondern dass er im Kontext funktioniert. Die Validierung umfasst oft Benutzerbewertungen, klinische Studien oder Tests unter simulierten Bedingungen, um Aspekte wie Benutzerfreundlichkeit, Sicherheit und Wirksamkeit aus Sicht der Endanwender abzudecken.
Die folgende Tabelle zeigt einige wichtige Unterschiede zwischen Verifikation und Validierung:
| Kriterium | Verifikation | Validierung |
|---|---|---|
| Zweck | Bestätigt, dass das Design die technischen Spezifikationen erfüllt. | Bestätigt, dass das Produkt den Benutzeranforderungen und dem Verwendungszweck entspricht. |
| Methoden | Inspektionen, Messungen, Labortests, Simulationen, Code-Reviews – Fokus auf technische Korrektheit. | Klinische Studien, Gebrauchstests, Leistungstests unter realen Bedingungen – Fokus auf Ergebnisse, Benutzerfreundlichkeit und Sicherheit. |
| Zeitpunkt | Während der Entwicklungsphase an Prototypen oder Komponenten durchgeführt, meist iterativ. | Gegen Ende der Entwicklung an finalen oder produktionsgleichen Geräten durchgeführt, unter realen Einsatzbedingungen. |
| Testobjekte | Kann an Prototypen, Mustern oder Teilsystemen erfolgen. | Muss an produktionsechten Geräten erfolgen, hergestellt mit finalen Fertigungsprozessen. |
| Fokus | Anforderungsgetrieben (wurden die Designvorgaben erfüllt?). Häufig intern (durch Entwicklerteams). | Verwendungsgetrieben (erfüllt es die Erwartungen der Nutzer?). Oft mit externen Beteiligten (z. B. Klinikpersonal), möglicherweise mit regulatorischer Prüfung. |
Sowohl Verifikation als auch Validierung sind verpflichtend – das eine kann das andere nicht ersetzen. Ein Gerät, das die Verifikation besteht, aber bei der Validierung scheitert, ist im Grunde die falsche Lösung (technisch korrekt, aber ungeeignet für den Anwender); umgekehrt kann ein Gerät zwar nützlich erscheinen, aber wenn es die Spezifikationen nicht zuverlässig erfüllt, ist es potenziell unsicher oder inkonsistent. Es ist absolut möglich, ein Gerät zu entwickeln, das alle technischen Anforderungen erfüllt, aber im praktischen Einsatz scheitert, weil es schwer zu bedienen ist oder das klinische Problem nicht tatsächlich löst. Aus diesem Grund verlangen Regulierungsbehörden weltweit, dass sowohl die Designverifikation als auch die Designvalidierung vor der Markteinführung durchgeführt werden. Die Verifikation schafft Vertrauen in die technische Korrektheit, und die Validierung gibt Sicherheit in Bezug auf Wirksamkeit und Sicherheit im realen Einsatz – gemeinsam liefern sie den Nachweis, dass ein Medizinprodukt sowohl richtig gebaut als auch die richtige Lösung ist.
Regulierungsbehörden in den Vereinigten Staaten und in Europa verlangen explizit robuste Verifikations- und Validierungsprozesse als Teil der Markteinführung von Medizinprodukten. Auch wenn sie unterschiedliche Terminologien und Schwerpunkte verwenden, fordern sowohl die FDA als auch die EU-MDR-Rahmenwerke, dass Hersteller durch dokumentierte Tests nachweisen, dass ihre Produkte den Spezifikationen entsprechen und die Anforderungen der Benutzer erfüllen.
In den USA verlangt die FDA von Herstellern medizinischer Geräte die Implementierung formeller Verifikations- und Validierungsprozesse im Rahmen ihrer Quality System Regulation (QSR). Gemäß 21 CFR 820.30 muss die Designverifikation bestätigen, dass die Designausgänge den Eingabeanforderungen entsprechen, während die Designvalidierung nachweisen muss, dass das Endgerät die Benutzeranforderungen und den vorgesehenen Gebrauch unter tatsächlichen oder simulierten Einsatzbedingungen erfüllt. Beide Aktivitäten müssen in der Design History File (DHF) dokumentiert werden. Die FDA legt besonderen Wert auf Rückverfolgbarkeit, produktionsechte Validierungstests und Usability-Validierung, insbesondere bei Software. Unzureichende V&V ist ein häufiger Grund für FDA-Warnschreiben, was die Einhaltung zu einem entscheidenden Faktor für die Marktzulassung macht.
In der EU verpflichtet die Medical Device Regulation (MDR) Hersteller, die Einhaltung der Allgemeinen Sicherheits- und Leistungsanforderungen (GSPR) nachzuweisen, was umfassende Verifikations- und Validierungsprozesse einschließt. Die Technische Dokumentation muss belegen, dass die Designausgänge (einschließlich Software) den Spezifikationen entsprechen und dass das Gerät klinisch sicher und wirksam ist. Die klinische Bewertung und Usability-Tests sind zentrale Elemente der Validierung, insbesondere bei risikoreichen oder patientennahen Produkten. Obwohl die MDR weniger konkret ist als die FDA, verlangt sie ein Qualitätsmanagementsystem (typischerweise nach ISO 13485), das V&V abdeckt, mit Anforderungen, die sich an Normen wie IEC 62304 (Software) und IEC 62366 (Usability) orientieren. Ohne ausreichende V&V-Nachweise wird keine CE-Kennzeichnung und kein Marktzugang gewährt.
Ergänzend zu den Vorschriften gibt es mehrere wichtige internationale Normen, die Best Practices für Verifikation und Validierung in der Entwicklung medizinischer Geräte definieren. Die Einhaltung dieser Normen ist oft freiwillig, wird jedoch nachdrücklich empfohlen (und ist in manchen Fällen faktisch verpflichtend – etwa wenn die EU eine Norm in die MDR aufnimmt oder die FDA eine Konsensnorm anerkennt). Nachfolgend ein Überblick über drei besonders relevante Normen: ISO 13485, ISO 14971 und IEC 62304 – sowie deren Zusammenhang mit V&V.
| Norm | Schwerpunkt |
|---|---|
| ISO 13485 | Norm für Qualitätsmanagementsysteme, die Designverifikation und -validierung als Teil der Entwicklung vorschreibt. |
| ISO 14971 | Risikomanagementnorm, in der Risikokontrollen verifiziert und die Gesamtsicherheit des Geräts validiert werden müssen. |
| IEC 62304 | Definiert Software-Lebenszyklusprozesse, einschließlich notwendiger Verifikation auf allen Ebenen der Softwareentwicklung. |
| IEC 62366-1 | Norm für Usability Engineering, die Verifikation und Validierung der Sicherheit der Benutzeroberfläche fordert. |
| IEC 60601-Serie | Normen zur elektrischen Sicherheit und elektromagnetischen Verträglichkeit mit Verifikationstests für medizinische Elektrogeräte. |
| ISO 10993 | Biologische Bewertungsnorm mit Anforderungen zur Verifikation der Biokompatibilität von Materialien. |
| ISO 11607 | Norm für Verpackung, die die Validierung der Integrität von sterilen Barrieren über Zeit und Transport hinweg fordert. |
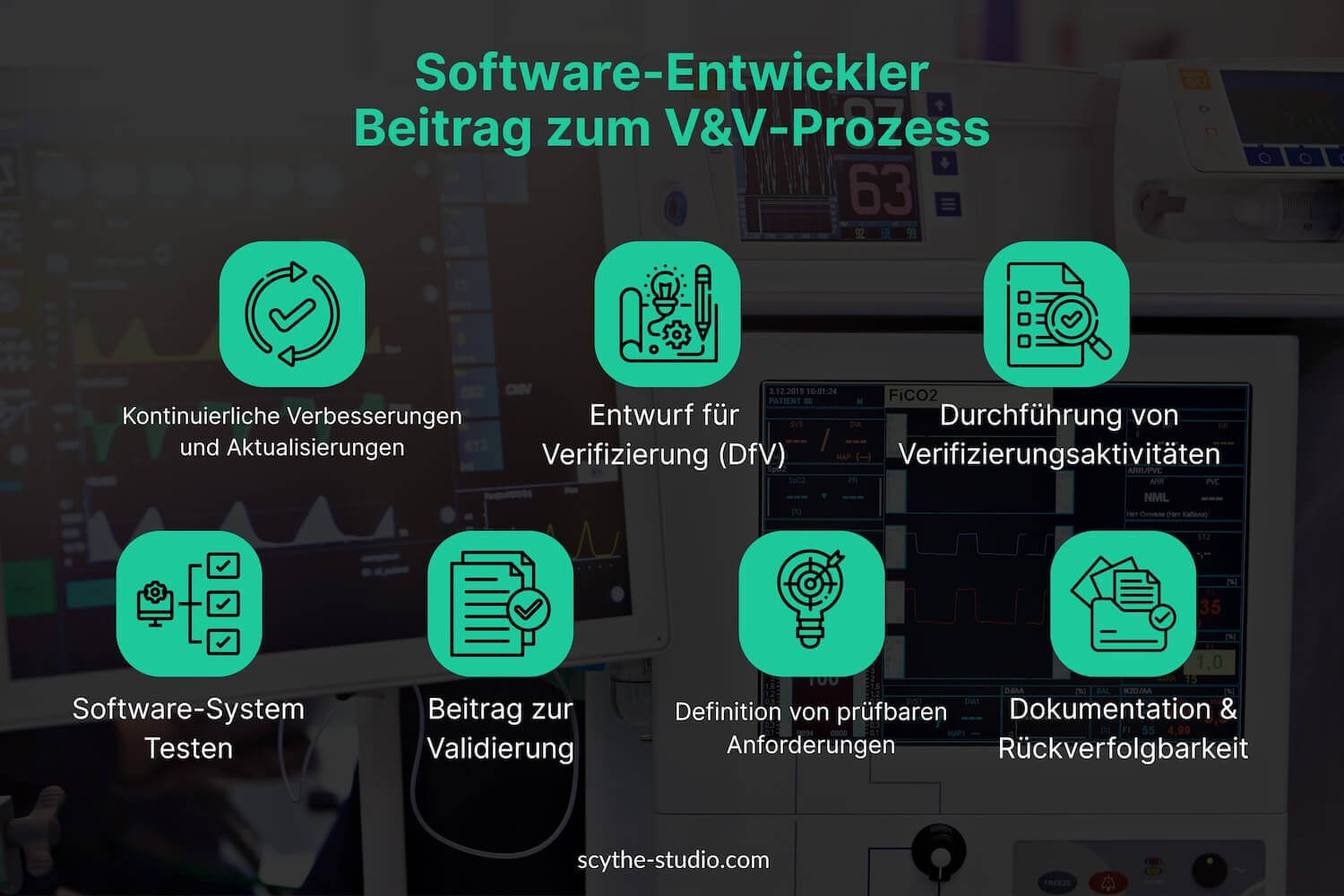
Moderne Medizinprodukte enthalten oft umfangreiche Softwarekomponenten – sei es eingebettete Firmware in Hardware oder eigenständige Software als Medizinprodukt (etwa eine mobile App mit diagnostischer Funktion). Deshalb spielen Softwareentwickler eine zentrale Rolle im V&V-Prozess. Anders als in manchen traditionellen Branchen, in denen das Testen ausschließlich dem QA-Team überlassen wird, arbeiten in der Medizintechnik die Softwareentwicklungsteams meist eng an Verifikations- und Validierungsaktivitäten mit. Hier einige Beiträge von Softwareingenieuren:
Zusammengefasst leisten Softwareentwickler Beiträge zu V&V nicht nur durch das Schreiben von Code, sondern auch durch aktive Beteiligung an dessen Verifikation und an der Produktvalidierung. Sie bringen das technische Know-how mit, um sicherzustellen, dass Tests gründlich und relevant sind. Ein starkes V&V-Team beinhaltet häufig Software-Spezialisten, die automatisierte Testscripte entwickeln, komplexe Protokolle auswerten und Sonderfälle erkennen können, die verifiziert werden müssen. Für Unternehmen, die in diesem Bereich Personal einstellen, sind Entwickler mit qualitätsorientierter Denkweise besonders wertvoll – nicht nur Codierer, sondern testbewusste Technologen.
Verifikation und Validierung umfassen eine Vielzahl von Testmethoden. Die genaue Methode hängt vom jeweiligen Gerätetyp ab – die V&V eines implantierbaren Herzschrittmachers unterscheidet sich stark von der einer Wellness-App. Im Folgenden stellen wir einige typische Testmethoden für eingebettete medizinische Software sowie für Mobile-Health-Anwendungen vor – mit einem Fokus darauf, wie sie die Ziele von V&V unterstützen.
Eingebettete medizinische Geräte (wie Infusionspumpen, Beatmungsgeräte, Diagnostikanalysatoren, implantierbare Geräte usw.) erfordern V&V auf Hardware- und Softwareebene – oft kombiniert.
Mobile-Health-Apps und andere reine Softwareprodukte (oft als Software as a Medical Device – SaMD bezeichnet) stellen besondere Testherausforderungen dar. Sie laufen in der Regel auf Standardhardware (z. B. Smartphones, Tablets) und können mit Cloud-Diensten oder Wearables integriert sein. Hier sind gängige V&V-Ansätze für solche Anwendungen:
All diese Tests und Analysen durchzuführen ist nur die halbe Miete – sie zu dokumentieren und die Rückverfolgbarkeit sicherzustellen ist im Bereich der Medizintechnik ebenso entscheidend. Regulierungsprüfer sagen oft: „Wenn es nicht dokumentiert ist, ist es nicht passiert.“ Deshalb ist ein zentrales Ergebnis von V&V eine umfassende Dokumentation darüber, was getestet wurde, wie es getestet wurde und welche Ergebnisse vorliegen – alles verknüpft mit den Designanforderungen und identifizierten Risiken.
Ein zentrales Werkzeug dafür ist der Validierungsplan. Eine Rückverfolgbarkeitsmatrix ist im Wesentlichen eine Tabelle (oder Datenbank), die Benutzeranforderungen → Designvorgaben → Designausgänge → Verifikationstests → Validierungstests miteinander verknüpft. Sie stellt sicher, dass es für jede Anforderung in jeder Phase entsprechende Verifikations- oder Validierungsnachweise gibt.
Dokumentation im V&V-Prozess umfasst in der Regel:
Ein Dokument, das früh in der Entwicklung erstellt wird und beschreibt, was verifiziert und validiert werden muss, welche Methoden angewendet werden, welche Anforderungen an die Testumgebung bestehen, wer verantwortlich ist usw. Es verweist häufig auf die Anforderungen und legt die Strategie fest (einschließlich Stichprobenbegründungen, z. B. wenn nicht jedes Gerät getestet wird). Regulierer erwarten, dass ein geplanter, strukturierter Ansatz vorliegt – kein improvisiertes Testen.
Ausführliche Anleitungen für jeden Test oder jede Testgruppe, in denen beschrieben wird, wie der Test durchzuführen ist, welches Equipment verwendet wird (einschließlich Kalibrierstatus), welche Akzeptanzkriterien gelten und welches die konkreten Schritte sind. Zum Beispiel enthält ein Protokoll zur „elektrischen Sicherheitsverifikation“ Verweise auf die Norm (60601-1), die konkreten Tests (Ableitstrom, Spannungsfestigkeit usw.) sowie die zulässigen Grenzwerte. Ein Software-Testprotokoll listet jede Testfall-ID mit den erwarteten Ergebnissen. Bei Software kann dies auch in einem Testmanagementsystem anstelle eines Word-Dokuments verwaltet werden – der Zweck bleibt derselbe.
Nach der Durchführung werden die Ergebnisse dokumentiert – typischerweise in einem Testbericht, der zusammenfasst, welche Tests bestanden oder nicht bestanden wurden, welche Abweichungen vorlagen, wer sie durchgeführt hat, Datum etc. Rohdaten oder Logdateien werden häufig als Anhang beigefügt. Fehlerhafte Tests führen zu Abweichungsberichten (Bugreports oder Non-Conformance Reports), die adressiert und erneut getestet werden. Diese Berichte dienen als objektiver Nachweis für die Verifikation.
Obwohl es sich nicht um Tests im engeren Sinne handelt, gehören Design Reviews (formelle Überprüfungen in verschiedenen Phasen) zur Designverifikation laut FDA. Sitzungsprotokolle oder unterzeichnete Berichte belegen, dass funktionsübergreifende Reviews (z. B. zu Anforderungen, Entwürfen, Code) durchgeführt und notwendige Maßnahmen umgesetzt wurden. Dies dokumentiert die analytische Verifikation durch fachliche Prüfung.
Für die Designvalidierung gibt es Protokolle für z. B. klinische Tests oder Usability-Studien (inkl. Teilnehmerzahl, Szenarien, Erfolgskriterien) sowie Berichte, die die Ergebnisse zusammenfassen (z. B. „95 % der Aufgaben wurden ohne Anwendungsfehler abgeschlossen – Erfolgskriterium erfüllt“). Bei klinischen Daten ist in der Regel ein Clinical Evaluation Report (CER) oder eine Zusammenfassung der klinischen Leistung Teil der Validierungsdokumentation.
Die Risikomanagementakte (gemäß ISO 14971) ist eng mit V&V verknüpft. Sie sollte die Verifikation jeder Risikokontrolle sowie die Validierung der Gesamtsicherheit enthalten. Häufig werden Testberichte in der Risikodatei referenziert, z. B.: „Risiko Überhitzung wird durch Temperatursensor mit Abschaltung bei 45 °C begrenzt – siehe Testbericht TR-123.“ Der Abschlussbericht beinhaltet in der Regel die Feststellung, dass alle Risikominderungsmaßnahmen verifiziert wurden und das Gesamtrisiko akzeptabel ist (was durch Nachweise zur sicheren Anwendung validiert wird).
Verifikations- und Validierungstests für Medizinprodukte sind ein anspruchsvolles Feld, das technisches Fachwissen, strukturierte Prozesse und einen geduldigen, gründlichen Ansatz erfordert. Richtig umgesetzt, sorgen sie dafür, dass lebensrettende und lebensverbessernde Technologien wirklich halten, was sie versprechen. Unternehmen, die in solide V&V-Praktiken und qualifizierte Fachkräfte investieren, bleiben nicht nur im Einklang mit den Anforderungen der FDA und EU-Verordnungen, sondern entwickeln auch Produkte von höherer Qualität – mit weniger Rückrufen und zufriedeneren Anwendern. In einer Branche, in der Vertrauen entscheidend ist, ist effektives V&V der Weg, es zu verdienen. Bauen Sie also Ihre Rückverfolgbarkeitsmatrizen, führen Sie gründliche Tests durch und vergessen Sie nie das eigentliche Ziel: ein sicheres und wirksames Medizinprodukt, validiert im Labor und bewährt im Einsatz.


Kommen wir zur Sache: Es ist eine Herausforderung, Top-Qt-QML-Entwickler zu finden. Helfen Sie sich selbst und starten Sie die Zusammenarbeit mit Scythe Studio – echten Experten im Qt C++ Framework.
Entdecken Sie unsere Fähigkeiten!Von Medizinprodukten und intelligenten Fahrzeugen bis hin zu Industriecontrollern und Unterhaltungselektronik – eingebettete Systeme sind allgegenwärtig und zunehmend vernetzt. Doch […]

Hey, willkommen zurück zu einem weiteren Blogbeitrag. Heute sprechen wir über das neue Qt WebAssembly. Dieser Beitrag hilft dir, die […]


Benutzer von eingebetteten Geräten – von Industriecontrollern bis hin zu Unterhaltungselektronik – sind sich oft nicht der versteckten Schwachstellen bewusst, […]