
Cybersecurity for Embedded Systems
From medical devices and smart vehicles to industrial controllers and consumer electronics, embedded systems are everywhere—and increasingly connected. But with […]

Join us at Qt C++ Warsaw Meetup - 21.08.2025
Sign up for free!
Verification and Validation (V&V) are two pillars that ensure a medical device is safe, effective, and compliant before it ever reaches patients. The verification process and validation process occur throughout the product development cycle – from initial design through production and post-market changes – and they serve as formal “checkpoints” to confirm quality and performance. In short, V&V is not just a regulatory box to tick – it’s a fundamental part of risk management, quality assurance and the testing process that protects users and companies alike, and ensures regulatory compliance.
In this article, you’ll learn:
At Scythe Studio, we support medical device teams in delivering high-quality, standards-compliant software through rigorous testing and validation. Whether you’re building embedded firmware or mobile health apps, we can help you strengthen your V&V testing medical device process and reduce regulatory risk, all while supporting efficient medical device design.
Let’s talk about how to make your next product safe, testable, and audit-ready.
AtMedical device companies and their customers have a lot at stake if V&V is not done right. Verification and validation provide confidence that a device will work correctly and safely throughout its life, directly impacting patient safety. Without a systematic process of checks against engineering device specifications and user requirements, developers cannot be certain that the finished product will:
In essence, V&V sits at the core of medical device industry quality standards and regulations because these activities catch design issues or performance gaps before they can harm patients or lead to costly field failures. The design validation process is crucial for confirming that a device performs as intended in real-world scenarios.
Verification and validation serve related but distinct purposes in ensuring device quality. Verification asks: “Did we build the product right?” – confirming the device is engineered correctly according to specifications. Validation asks: “Did we build the right product?” – confirming the finished device meets user needs and performs as intended in real-world use. Both aspects are essential. A device might pass all verification tests (meeting every technical spec) yet fail validation if it doesn’t solve the user’s actual problem or is unsafe or impractical in the field. Thorough V&V, therefore, is how manufacturers prevent costly redesigns, reduce regulatory risk, and ensure devices are reliable for real-world medical use. Well-documented test results are essential for demonstrating compliance and supporting future audits.
From a lifecycle perspective, V&V activities are woven into the design process as formal checkpoints (often tied to design controls). Verification often occurs iteratively during development – testing medical device prototypes or components against design inputs – while validation occurs on production-equivalent devices (or final design) to prove the device works for the end-user. Verification occurs early in the development phase, whereas validation is often done later, on the final production units. Verification determines whether design outputs match device specifications. Validation determines if the product meets its intended use.
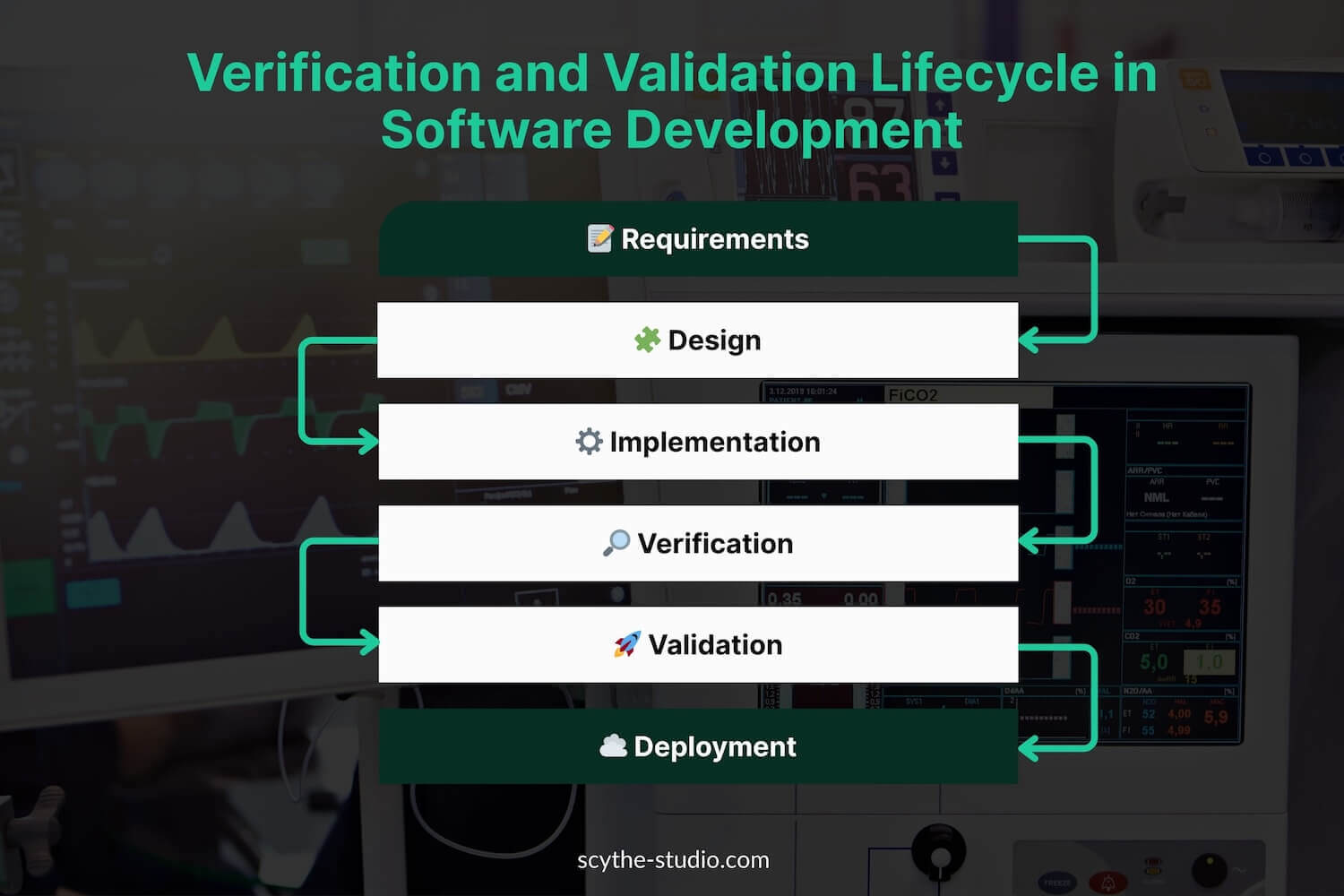
Regulatory agencies require evidence of both before a product can be cleared or approved for market. In the U.S., the FDA will not clear a device (510(k), PMA, etc.) without documentation that design outputs were verified against inputs and that the final device was validated to meet user needs. In Europe, Notified Bodies reviewing a Technical File under EU MDR expect to see comprehensive V&V results (including clinical evidence) showing compliance with the General Safety and Performance Requirements. In short, V&V is the quality gate that every medical device must pass to ensure it’s not just built correctly, but is the correct solution for patients.
It’s easy to confuse verification and validation because both involve testing and produce objective evidence. However, they answer fundamentally different questions. A classic way to remember is:
In formal terms, the FDA and ISO definitions reflect this distinction. Design verification is defined as “confirmation by examination and provision of objective evidence that specified requirements have been fulfilled”. In practice, those “specified requirements” are the design input requirements – the engineering specifications derived from user needs. Verification activities answer the question: Did we correctly implement each requirement in the design? For example, if a design input states “the catheter shall withstand at least 200 psi burst pressure,” verification would entail testing the catheter’s burst pressure to confirm it meets that spec. Every design output (drawings, prototypes, code, etc.) is checked to ensure it conforms to the input requirements.
Design validation, on the other hand, is “confirmation by examination and provision of objective evidence that the particular requirements for a specific intended use can be consistently fulfilled”. Less formally, validation asks: Does the device, as built, satisfy the user needs and intended medical use in the real world? This involves testing the final product (or production-quality units) under actual or simulated use conditions to ensure the device is fit for its intended purpos. For instance, validating that a catheter actually improves patient outcomes in a clinical setting and is usable by clinicians – not just that it meets engineering specs, but that it performs in context. Validation often includes user evaluations, clinical studies, or simulated use testing to cover aspects like usability, safety, and efficacy in the hands of end-users.
The table below highlights some key differences between verification and validation:
| Criterion | Verification | Validation |
|---|---|---|
| Purpose | Confirms the design meets technical specifications. | Confirms the product meets user needs and intended use. |
| Methods | Inspections, measurements, bench tests, simulations, code reviews – focused on technical accuracy. | Clinical trials, usability studies, performance tests in real-world scenarios – focused on outcomes, usability, and safety. |
| Timing | Conducted throughout development process on prototypes or components, during design iterations. | Performed near the end of development on final or production-equivalent units, under real-world conditions. |
| Test Articles | Can be performed on prototypes, samples, or sub-systems. | Must be conducted on production-equivalent devices built with final manufacturing processes. |
| Focus | owspan=”1″>Requirements-driven (did we meet design input specifications?). Often internal (by engineering teams). | Use-case-driven (does it meet user expectations?). Often involves external stakeholders (e.g., clinicians), may require regulatory approval. |
Both verification and validation are mandatory – one cannot replace the other. A device that passes verification but fails validation is essentially the wrong solution (meets specs but not user needs); conversely, one that somehow seems useful but doesn’t meet specs reliably could be inconsistent or unsafe. It is entirely possible to design a device that meets all its engineering requirements yet flops in the field because it’s hard to use or doesn’t actually solve the clinical problem. That’s why regulations worldwide insist on doing both design verification and design validation prior to market release. Verification gives confidence in technical correctness, and validation gives confidence in real-world effectiveness and safety – together they provide the evidence that a medical device is both built right and the right solution.
Regulatory bodies in the United States and Europe explicitly require robust V&V processes as part of bringing a medical device to market. While they use slightly different terminology and emphasis, both FDA and EU MDR frameworks demand that manufacturers prove their devices meet specifications and fulfill user needs through documented testing.
In the U.S., the FDA requires medical device manufacturers to implement formal verification and validation processes as part of their Quality System Regulation (QSR). Under 21 CFR 820.30, design verification must confirm that design outputs meet input requirements, while design validation must demonstrate that the final device meets user needs and intended uses under actual or simulated use conditions. Both activities must be documented in the Design History File (DHF). FDA places strong emphasis on traceability, production-equivalent validation testing, and usability validation, especially for software. Inadequate V&V is a common reason for FDA warning letters, making compliance critical for market approval.
In the EU, the Medical Device Regulation (MDR) mandates that manufacturers demonstrate compliance with General Safety and Performance Requirements (GSPR), which inherently includes robust verification and validation. Technical Documentation must show evidence that design outputs (including software) meet specifications and that the device is clinically safe and effective. Clinical evaluation and usability testing are central to validation, especially for higher-risk or patient-facing devices. While MDR is less prescriptive than the FDA, it requires a quality system (typically ISO 13485) that covers V&V, with expectations aligned to standards like IEC 62304(software) and IEC 62366 (usability). Without sufficient V&V evidence, CE marking and market access are not granted.
Overlaying the regulations are several key international standards that define best practices for V&V in medical device development. Compliance with these standards is often voluntary but strongly encouraged (and sometimes essentially required, as when the EU “harmonizes” a standard to MDR or the FDA recognizes a consensus standard). Let’s look at three of the most relevant standards: ISO 13485, ISO 14971, and IEC 62304 – as well as how they interact with V&V.
| Standard | Focus |
|---|---|
| ISO 13485 | Quality management system standard requiring design verification and validation as part of development. |
| ISO 14971 | Risk management standard where risk controls must be verified and overall device safety validated. |
| IEC 62304 | Defines software lifecycle processes, including required verification of software at all levels. |
| IEC 62366-1 | Usability engineering process requiring verification and validation of user interface safety. |
| IEC 60601 series | Electrical safety and EMC standards defining verification tests for medical electrical equipment. |
| ISO 10993 | Biological evaluation standard requiring verification of material biocompatibility. |
| ISO 11607 | Packaging standard requiring validation of sterile barrier system integrity over time and transport. |
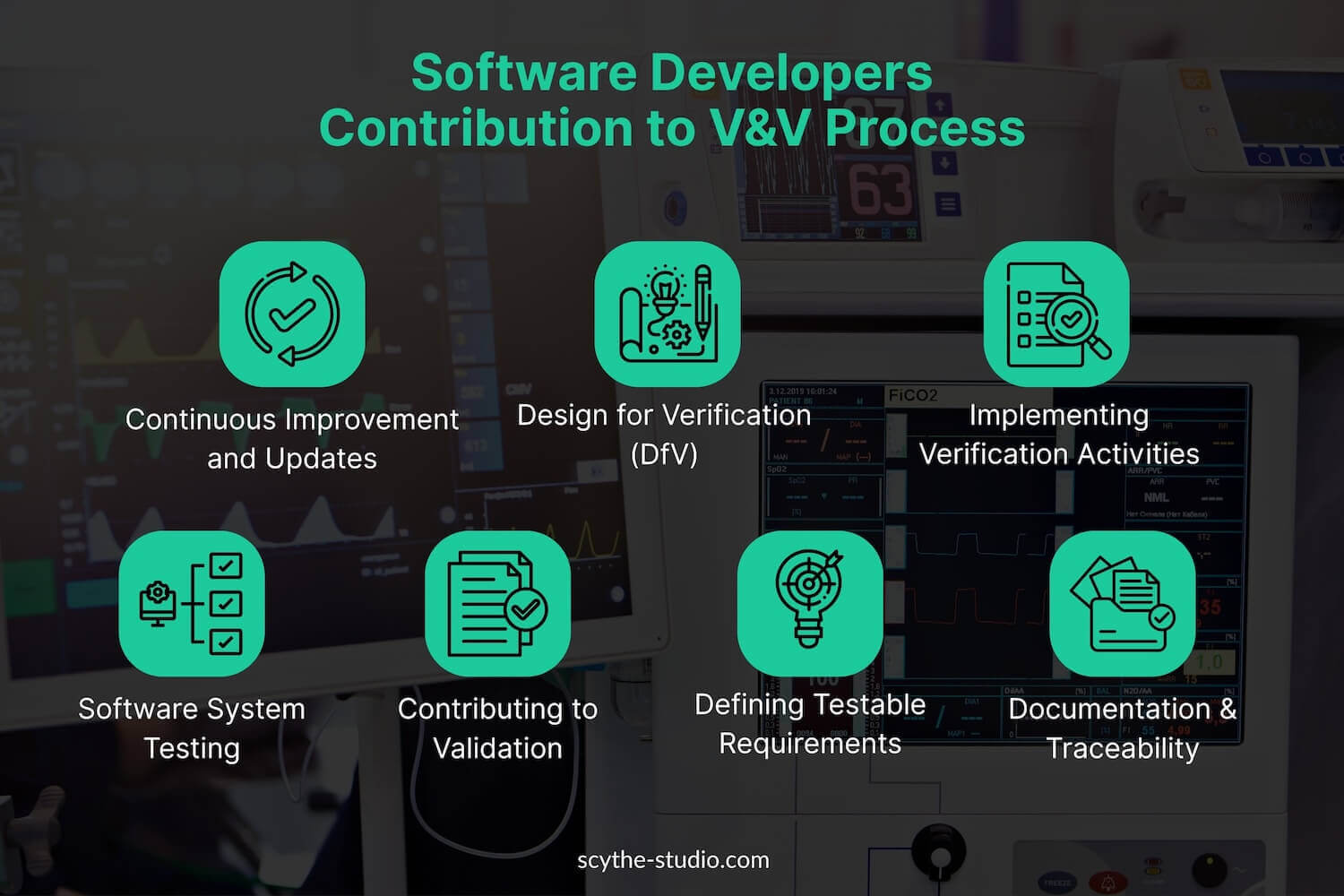
Modern medical devices often have significant software components – whether it’s embedded firmware in hardware or standalone software as a medical device (like a mobile app that performs diagnostic functions). As a result, software engineers play a crucial part in the V&V process. Unlike some traditional industries where testing might be left solely to a QA department, in medical devices the software development team typically collaborates closely in verification and validation activities. Here’s how software engineers contribute:
In summary, software engineers contribute to V&V not only by writing code but by actively participating in the verification of that code and validation of the product. They bring technical expertise that ensures tests are thorough and relevant. A strong V&V team often includes software specialists who can develop automated test scripts, interpret complex data logs, and anticipate edge-case scenarios that require verification. For organizations hiring in this area, it’s valuable to have software engineers who are not just coders, but test-minded developers with a quality-first mentality.
Verification and validation encompass a wide array of testing methods. The exact methods used will depend on the type of device – for instance, an implanted pacemaker’s V&V differs from a wellness mobile app’s V&V. Here we outline some common testing methods for embedded medical device software and for mobile health applications, highlighting how they support V&V goals.
Embedded medical devices (such as infusion pumps, ventilators, diagnostic analyzers, implantable devices, etc.) require V&V at both the hardware and software level, often in combination.
Mobile health apps and other software-only medical devices (often categorized as Software as a Medical Device, SaMD) present some different testing challenges. They typically run on general-purpose hardware (e.g., smartphones, tablets) and might integrate with cloud services or wearable sensors. Here are common V&V approaches for these:
Running all these tests and analyses is only half the battle – documenting them and maintaining traceability is equally important in the medical device arena. Regulatory auditors often say, “If it’s not documented, it didn’t happen.” Thus, a key output of V&V is a comprehensive set of documents showing what was tested, how it was tested, and the results, all linked back to the design requirements and risks.
A central tool for this is the validation plan. A traceability matrix is essentially a table (or database) that links user needs → design inputs → design outputs → verification tests → validation tests. It ensures that for every requirement at each stage, there is corresponding evidence of verification or validation.
Documentation in V&V typically includes:
A document prepared early in development describing what needs to be verified and validated, the methods to be used, test environment requirements, responsibilities, etc. It often references the requirements and outlines the strategy (including any sampling rationale, e.g. if not every unit is tested). Regulators like to see that there was a planned approach, not ad-hoc testing.
Detailed documents for each test or group of tests explaining how to perform the test, what equipment to use (including calibration status), acceptance criteria, and the step-by-step procedure. For example, a protocol for “electrical safety verification” would cite the standard (60601-1), list the specific tests (earth leakage, dielectric strength, etc.), and the passing thresholds. Similarly, a software test protocol might list each test case and expected result. In some cases, especially for software, these test cases may reside in a test management tool rather than a Word document, but they serve the same purpose.
After execution, results are recorded. This can be in the form of a test report summarizing all outcomes (which tests passed, which failed, any deviations, who performed it, date, etc.), often attaching raw data or logs. Any failed tests should trigger issue reports (bug reports or non-conformance reports) that get addressed and re-tested. These reports demonstrate objective evidence of verification.
Although not a test, design reviews (formal reviews at various stages) are part of design verification per FDA. Meeting minutes or signed records show that cross-functional reviews were conducted (for requirements, designs, code, etc.) and any actions were resolved. This is documentation that the design was scrutinized – a form of verification via analysis.
For design validation, you’ll have protocols for things like clinical testing or usability studies (describing how many users, what scenarios, success criteria) and then reports that compile the results (e.g., “95% of tasks were completed without use error in the study, meeting the success criteria”). If clinical data is used, typically a Clinical Evaluation Report (CER) or summary of clinical performance is part of validation documentation.
The risk management file (from ISO 14971 process) is tied in as well. It should contain verification that each risk control was implemented and effective. Often, test reports will be referenced in the risk file to show, for example, “risk of overheating mitigated by temp sensor shutdown – see Test Report TR-123 confirming shutdown at 45°C.” The final report often states that all risk control measures have been verified and that overall residual risk is acceptable (which is effectively validated by the evidence that the device is safe in use).
Verification and validation testing for medical devices is a challenging domain that requires technical expertise, rigorous process, and a patient, thorough approach. But done right, it ensures that life-saving and life-improving technologies truly deliver on their promises. Companies that invest in strong V&V practices – and the talented people to lead them – will not only stay compliant with FDA and EU regulations, but will also build devices of higher quality with fewer recalls and happier users. In an industry where trust is paramount, effective V&V is how you earn it. So, build those traceability matrices, run those exhaustive tests, and never forget the end goal: a safe and effective medical device that improves lives, validated in the lab and proven in the field.


Let's face it? It is a challenge to get top Qt QML developers on board. Help yourself and start the collaboration with Scythe Studio - real experts in Qt C++ framework.
Discover our capabilitiesFrom medical devices and smart vehicles to industrial controllers and consumer electronics, embedded systems are everywhere—and increasingly connected. But with […]

Hey, welcome back to another blog post. Today we’re going to talk about the new Qt WebAssembly. This post will […]


Users of embedded devices – from industrial controllers to consumer electronics – are often unaware of hidden vulnerabilities that threaten […]